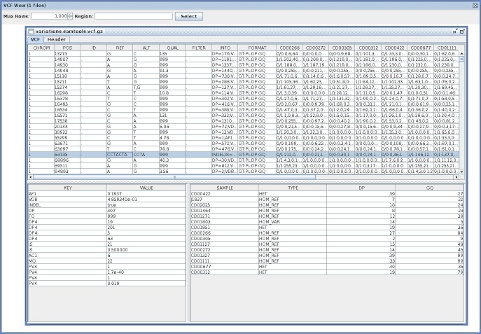
A quick note about three java-based tools for VCF files I wrote today.
VcfViewGui
VcfViewGui : a Simple java-Swing-based VCF viewer.
VCFGeneOntology
vcfgo reads a VCF annotated with
VEP or
SNPEFF, loads the data from
GeneOntology and
GOA and adds a new field in the INFO column for the GO terms for each position.
Example:
$ java -jar dist/vcfgo.jar I="https://raw.github.com/arq5x/gemini/master/test/tes.snpeff.vcf" |\
grep -v -E '^##' | head -n 3
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT 1094PC0005 1094PC0009 1094PC0012 1094PC0013
chr1 30860 . G C 33.46 . AC=2;AF=0.053;AN=38;BaseQRankSum=2.327;DP=49;Dels=0.00;EFF=DOWNSTREAM(MODIFIER||||85|FAM138A|protein_coding|CODING|ENST00000417324|),DOWNSTREAM(MODIFIER|||||FAM138A|processed_transcript|CODING|ENST00000461467|),DOWNSTREAM(MODIFIER|||||MIR1302-10|miRNA|NON_CODING|ENST00000408384|),INTRON(MODIFIER|||||MIR1302-10|antisense|NON_CODING|ENST00000469289|),INTRON(MODIFIER|||||MIR1302-10|antisense|NON_CODING|ENST00000473358|),UPSTREAM(MODIFIER|||||WASH7P|unprocessed_pseudogene|NON_CODING|ENST00000423562|),UPSTREAM(MODIFIER|||||WASH7P|unprocessed_pseudogene|NON_CODING|ENST00000430492|),UPSTREAM(MODIFIER|||||WASH7P|unprocessed_pseudogene|NON_CODING|ENST00000438504|),UPSTREAM(MODIFIER|||||WASH7P|unprocessed_pseudogene|NON_CODING|ENST00000488147|),UPSTREAM(MODIFIER|||||WASH7P|unprocessed_pseudogene|NON_CODING|ENST00000538476|);FS=3.128;HRun=0;HaplotypeScore=0.6718;InbreedingCoeff=0.1005;MQ=36.55;MQ0=0;MQRankSum=0.217;QD=16.73;ReadPosRankSum=2.017 GT:AD:DP:GQ:PL 0/0:7,0:7:15.04:0,15,177 0/0:2,0:2:3.01:0,3,39 0/0:6,0:6:12.02:0,12,143 0/0:4,0:4:9.03:0,9,119
chr1 69270 . A G 2694.18 . AC=40;AF=1.000;AN=40;DP=83;Dels=0.00;EFF=SYNONYMOUS_CODING(LOW|SILENT|tcA/tcG|S60|305|OR4F5|protein_coding|CODING|ENST00000335137|exon_1_69091_70008);FS=0.000;GOA=OR4F5|GO:0004984&GO:0005886&GO:0004930&GO:0016021;HRun=0;HaplotypeScore=0.0000;InbreedingCoeff=-0.0598;MQ=31.06;MQ0=0;QD=32.86 GT:AD:DP:GQ:PL ./. ./. 1/1:0,3:3:9.03:106,9,0 1/1:0,6:6:18.05:203,18,0
VCFFilterGeneOntology
vcffiltergo reads a VCF annotated with
VEP or
SNPEFF, loads the data from
GeneOntology and
GOA and adds a filter in the
FILTER column if a gene at the current genomic location is a descendant of a given GO term.
Example:
$ java -jar dist/vcffiltergo.jar I="https://raw.github.com/arq5x/gemini/master/test/test1.snpeff.vcf" \
CHILD_OF=GO:0005886 FILTER=MEMBRANE |\
grep -v "^##" | head -n 3
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT 1094PC0005 1094PC0009 1094PC0012 1094PC0013
chr1 30860 . G C 33.46 PASS AC=2;AF=0.053;AN=38;BaseQRankSum=2.327;DP=49;Dels=0.00;EFF=DOWNSTREAM(MODIFIER||||85|FAM138A|protein_coding|CODING|ENST00000417324|),DOWNSTREAM(MODIFIER|||||FAM138A|processed_transcript|CODING|ENST00000461467|),DOWNSTREAM(MODIFIER|||||MIR1302-10|miRNA|NON_CODING|ENST00000408384|),INTRON(MODIFIER|||||MIR1302-10|antisense|NON_CODING|ENST00000469289|),INTRON(MODIFIER|||||MIR1302-10|antisense|NON_CODING|ENST00000473358|),UPSTREAM(MODIFIER|||||WASH7P|unprocessed_pseudogene|NON_CODING|ENST00000423562|),UPSTREAM(MODIFIER|||||WASH7P|unprocessed_pseudogene|NON_CODING|ENST00000430492|),UPSTREAM(MODIFIER|||||WASH7P|unprocessed_pseudogene|NON_CODING|ENST00000438504|),UPSTREAM(MODIFIER|||||WASH7P|unprocessed_pseudogene|NON_CODING|ENST00000488147|),UPSTREAM(MODIFIER|||||WASH7P|unprocessed_pseudogene|NON_CODING|ENST00000538476|);FS=3.128;HRun=0;HaplotypeScore=0.6718;InbreedingCoeff=0.1005;MQ=36.55;MQ0=0;MQRankSum=0.217;QD=16.73;ReadPosRankSum=2.017 GT:AD:DP:GQ:PL 0/0:7,0:7:15.04:0,15,177 0/0:2,0:2:3.01:0,3,39 0/0:6,0:6:12.02:0,12,143 0/0:4,0:4:9.03:0,9,119
chr1 69270 . A G 2694.18 MEMBRANE AC=40;AF=1.000;AN=40;DP=83;Dels=0.00;EFF=SYNONYMOUS_CODING(LOW|SILENT|tcA/tcG|S60|305|OR4F5|protein_coding|CODING|ENST00000335137|exon_1_69091_70008);FS=0.000;HRun=0;HaplotypeScore=0.0000;InbreedingCoeff=-0.0598;MQ=31.06;MQ0=0;QD=32.86 GT:AD:DP:GQ:PL ./. ./. 1/1:0,3:3:9.03:106,9,0 1/1:0,6:6:18.05:203,18,0
That's it,
Pierre